Baby Maverick defies expectation in blood cancer battle
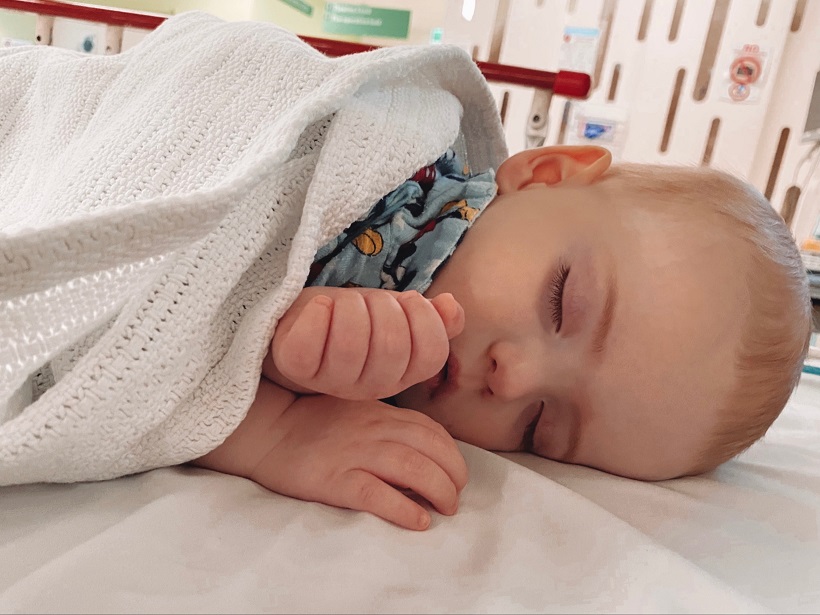
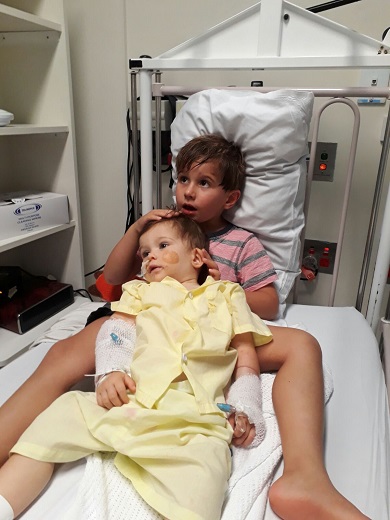
At only nine-months-old, Maverick Hill was diagnosed with a life-threatening blood cancer and forced to endure immediate and aggressive treatment away from home.
Maverick was only nine-months-old when he was diagnosed with leukaemia.
Maverick’s mum, Georgia remembers how suddenly he started to show symptoms.
“In early October 2020, we were still in lockdownin our hometown, Balnarring which is an hour and a half south of Melbourne,” said Georgia.
The Hill family celebrating Maverick’s 1st birthday in January 2021.
“SoI rang the doctor when Mav started spiking fevers and then had a couple of blood noses overnight.
“The nurse who was on callsaid not to worry, that it was probablyviral infection but to call again in a few days if his symptomspersist.”
Within days, Maverick started to develop a rash and bruising which would notblanchunder pressure.
Georgia called the doctor again who suspected meningitis and said to bring him in after the weekend if he did not improve.
“The doctor took one look at him on Monday and said he needed to have blood tests immediately,” said Georgia.
“I remember the Wednesday morning I got the callabout his results…I had just dropped my older daughter at daycare and was buying a rack of ribs at the butcher.
“Igrabbed my husband, Brodie and Mav and we raced into the surgery where we were told to go straight to the children’s hospital in Melbourne.”
After what Georgia considers ‘the longest drive of our lives’, the family arrived at the hospital and was put through a number of routine tests.
“It was all a huge blur but eventually the oncology team came down and broke the news to us that Maverick had leukaemia,” said Georgia.
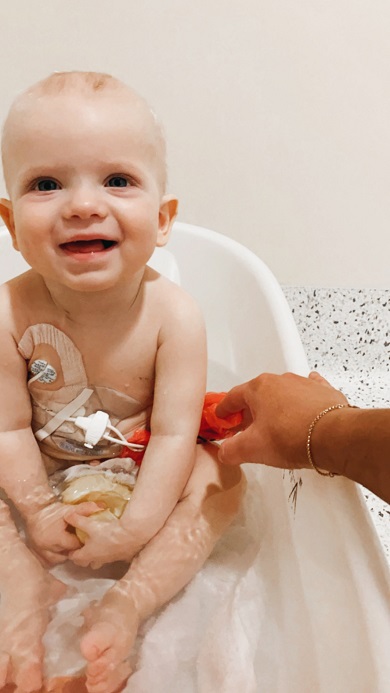
Maverick has had an incredible response to treatment so far.
“We barely had time to process the news before they started preparing him for a lumbar puncture and bone marrow biopsy.
“He was beside himself by this time, hadn’t slept and had been poked and prodded as they couldn’t find his vein to draw blood.
“They had to put both of his arms in splints to keep them straight so they could get cannulas in.
“We had to pin him down and he wouldn’t stop screaming – it was heartbreaking”
“While an ALL diagnosis is rare for infants under 12–months–old, Mav’s results cameback showing that his particular disease was low-risk and entirely treatable,” explained Georgia. “That was a huge relief.”
“We were incredibly grateful, as we had heard the accommodation had been really busy and Brodie had been doing the three-hour round trip back homeevery day,” said Georgia.
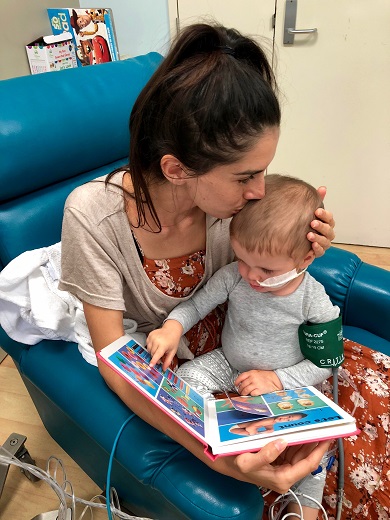
Maverick and sister, Mackenzie still got into the Christmas spirit during his treatment.
“That‘s the last thing you want to do after spending the whole day in hospital with your very sick baby.
“The unit is only a 10-minute walk from the hospital, we’ve made it our little ‘city home’and being able to stay altogether has been really important.”
Maverick has responded beyond expectation to his treatment, achieving remission after two weeks on steroids so he could begin chemotherapy.
“He reacted really well to the steroids, which was amazing, and we have now almost completed his induction stage of chemotherapy,” explained Georgia.
“We were warned that he was going to be really unwell throughout treatment and to prepare for the fevers, loss of appetite, nausea and fatigue.
“But he was incredible and practically did the opposite of anything they said he was going to do.
Maverick recently got to hang out with his close cousin, Jordi for the first time in 6 months.
“Although, the steroids did make him super angry and really hungry. He stopped sleeping and they made his face quite fat.
“During the third cycle he also got a bad case of mucositis.But that was only really for a week and the consultant said he had one of the fastest recovering bone marrows they’d seen.”
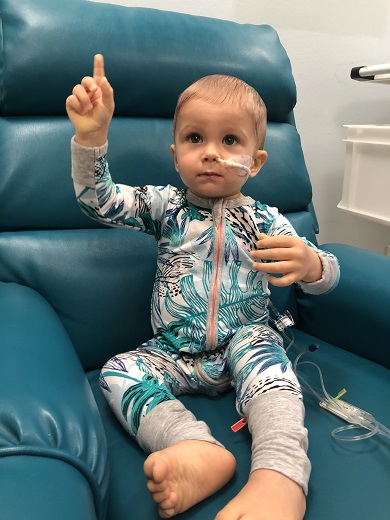
Whilethe start to Maverick’s treatment has been largelypositive, the family understand their blood cancer journey is far from over with more chemotherapy and then 18-months of maintenance treatment ahead.
“From the beginning, we’ve said we’ll just take it one step at a time and take each day as it comes,” said Georgia.
“We don’t like to overwhelm ourselves with everything that’s to comebut just live in the moment and be there for Mav as best we can.
“We are beyond grateful to have a great support network of family, friends and the Leukaemia Foundation behind us.
“We couldn’t imagine going through this without it.”
To mark Rare Disease Day on February 28, Laura Winter shares the remarkable story of how her four-year-old son, Flynn, has survived an extremely rare blood cancer diagnosis.
In late 2017, the Winter family, including dad, James, and seven-year-old big brother, Xavier, were living a blissful and simple life on tropical Christmas Island.
The Winter Family.
“Flynn then started to get rashes that would come and go, a bloated stomach, and would often get colds which would last a lot longer than his brother,” explained Laura.
“I thought it was all a bit off, but I never even considered leukaemia.”
When Flynn came down with pneumonia, a blood test finally revealed the devastating truth and the family needed to be airlifted to Perth to start urgent treatment.
“We had to leave our home, our friends and our life behind almost immediately to begin the fight of Flynn’s life. It was like a rug was pulled out from underneath us,” said Laura.
Flynn had a life-saving transplant at just 18 months old.
“JMML is incredibly rare with one in 1.2 million diagnosed,” explained Laura.
“I’m an avid researcher and when we received the diagnosis, I spent hours and hours trying to understand everything I could about JMML.
“Getting a rare diagnosis can be very isolating and uncertain as the research, treatment and statistics are changing all the time.
“The treatment team was, of course, very good at explaining everything, but, in the end, we only had one option: Flynn would need a bone marrow transplant.”
Flynn had a bone marrow transplant at just 18 months old from an unrelated donor as his brother wasn’t a match.
Flynn with big brother, Xavier.
Even then, the road forward for Flynn was complicated as he developed graft versus host disease (GVHD) within weeks of the transplant, which attacked his liver, skin, and gut. GVHD is common, and sometimes serious, complication of a transplant, where some of the donor’s immune cells try to reject the patient’s own cells as foreign.
“We pretty much lived in the hospital for most of that year after the transplant,” remembered Laura.
“He’s been on a huge journey with so many ups and downs but he’s a very resilient, amazing boy.”
Because of Flynn’s age, Laura says it is unlikely he had any concept of what was happening to him throughout that year.
“He couldn’t have been thinking about the next day, or what was going to happen to him,” said Laura.
Laura and Flynn during his treatment.
“Although, because he was so young, it was also a problem because we couldn’t explain what was happening.
“We just tried our best to keep him active, happy and smiling through it all and focus on the positives.
“The side effects were so horrific, but, for him, it was really just one day at a time.”
For the rest of the family, grappling with the realities of Flynn’s diagnosis was deeply affecting and they too focused on taking each day at a time and learning from others who had been through the same thing.
“Whatever would help us on that day, we’d find it,” said Laura.
“I got in contact with other JMML parents across the world, finding online support groups to ask questions and make me feel less alone.”
“I know first hand that when you’re in the midst of it, it’s really hard to find hope some days, so to hear these stories and to see others who are on the other side of it all can be really inspirational.”
Flynn kept smiling by mum and dad.
Flash forward to today, the family has just celebrated three years since Flynn’s transplant.
He now looks like any other cheerful, bubbly, and healthy four-year-old. Although he was unaware of the severity of his circumstances at the time, today Flynn is very aware of how special he is.
Now thriving, he doesn’t quite understand why he needs to go back to get his blood tests every two months.
“He says, ‘Why am I getting this test, Mum? Why am I going to hospital?’ and I just tell him, ‘To show how strong and golden your super blood is!’”
Life for the Winter family might have been transformed forever, but Laura’s confident that Flynn can grow up and experience life like any other kid.
“When he was immunocompromised, he wasn’t allowed to go to playgrounds or playgroups as any infection or bacteria could be a deadly threat,” said Laura. “He was very much in isolation at home for a good year.
Flynn has just started kindy this year and is thriving.
“But now, life is as normal as it can be as he continues to regain his strength and heal his body. We’ve stayed in Perth for the time being and he’s started kindy this year. We are so grateful.”
The family are now passionate about raising awareness of JMML and hope to see an improvement in future research and support.
“It is difficult with rare diseases to get funding for research and make real progress with new treatments,” said Laura.
“With JMML still only having about a 50% cure rate, the beautiful future children diagnosed deserve better outcomes.
“My hope for the future is that increased research would lead to targeted therapies, so that not every child would need to go to transplant to be cured.
“To potentially be able to avoid such a toxic procedure would be a huge step forward.”
Laura’s advice to others with a rare disease diagnosis:
Don’t be afraid to ask questions. I found it really empowering. Don’t think of it like you’re questioning your specialist’s authority, you are just getting it clear in your head for you and your child. Be sure to lean on others for support, and always, always keep hope.
Expert Series: Dr Cecily Forsyth on how to live your best life with MPN
Dr Cecily Forsyth and the nurse practitioner Jacqui Jagger
Dr Cecily Forsyth talks to her MPN patients “a lot” about what they can do to improve their health, often highlighting the findings of a study on integrative medicine in MPN.
According to the results, published last year, of an international survey of 858 participants, integrative medicine offers unique symptom management strategies. These are based on interventions such as aerobic activity, strength training and massage for decreased MPN symptom burden, depression, fatigue, and higher overall quality of life.
“The SIMM study was coordinated by Ruben Mesa*, who is the king of MPN symptoms,” said Dr Forsyth, a clinical haematologist with a special interest in the MPNs.
“The survey showed that when you’re living with a chronic malignancy you can improve your symptoms by the things you do, not just the pharmacotherapy that your haematologist prescribes.
“Integrating physical activity, yoga, massage, art therapy–all kinds of different things–can improve your quality of life,” Dr Forsyth explained.
“This is an important message; it’s all about how to live your best life.”
Dr Cecily Forsyth in the Lofoten Islands, Norway
Looking at the information and discussion in some posts on some Facebook support groups for MPN, Dr Forsyth felt there was too much negativity.
“Giving people a different, optimistic, message is really important,” she said.
“It’s about asking yourself, how do I live my best life, and how do I manage my symptoms so I can live my best life?”
“Helping patients with that is incredibly important, and it’s really well received; patients like to know that they can do things that can improve their symptoms.
“We know MPNs cause a lot of symptoms, but I don’t get bogged down about what’s causing them,” said Dr Forsyth.
“Whether your fatigue is from your MPN, stressful job, grouchy husband, or difficult children is irrelevant? What is relevant is that you are tired and how can we help you manage your fatigue to the best of our ability?
“It’s about moving your focus, because there’s no whizzbang therapy that means suddenly your symptoms have gone.
“It’s a way of managing them, and that will include patient-initiated activities and strategies,” said Dr Forsyth, whose nurse practitioner, Jacqui Jagger, also has an interest in symptom management in MPN.
“It’s about making sure you’re productive, you still have good relationships in your life, and you still have financial, work and vocational goals that all contribute to having a good quality of life. We try and work with our patients to achieve this.”
The SIMM study showed people do “a wide variety of activities” to try and help improve their lives, and those who exercise and who control their weight do have improvement in their symptoms.
“I explain to patients that they can try a wide range of activities such as yoga, strength training, Tai Chi, dance therapy …” and find what they enjoy and what inspires them.
“I tell patients repeatedly, exercise is needed. It’s absolutely fine to have a lovely massage but you also need to address the exercise and activity side of integrative medicine as well’.
“Participating in any kind of physical activity and strength training will improve your symptoms; you need to find an activity that suits you.
“I’m not a runner, I don’t enjoy that. But I’ve taken up pilates in the last five years, which I really enjoy.
“And I tell patients… that’s my time, when I’m not thinking about what work I haven’t completed? What am I going to cook for dinner? Have I done the washing? When am I going to do the letters?
Dr Cecioly Forsyth “planking” on the salt flats in Bolivia
“I’m too busy thinking, ‘how on earth am I going to get my leg up that high?”
“And I tell them that it clears the white noise out of my head. I can’t think about the stresses of work and life in a pilates class. I started pilates for my back but have found it to be just as good for my head as my back!
“I often give that example to patients and say, ‘you need to find what’s right for you’, whether it’s meditation or having a massage–things that allow you to destress.
“Doing an activity where you have to concentrate and focus on that activity, it could be art or ceramics, allows you to escape the worry about your MPN and life’s stresses.
“All this can help, particularly our patients with polycythaemia vera (PV) and essential thrombocythaemia (ET),” said Dr Forsyth.
“We want to try and make sure they’re in the healthy weight range, that they don’t smoke, and their cholesterol is under control… the whole range of interventions that will have an impact on symptom burden, quality of life, and risk of complications from the disease.”
Video consults and second opinions
Since the pandemic hit in 2020, Dr Forsyth has become an advocate of video call consultations and she’s convinced “they’re fabulous”.
“It’s one of the things that’s really improved the service for patients,” she said.
“I’m surprised by how useful they’ve been and, in the MPN patient population, it’s enabled people to see a haematologist with a little bit more of an interest in MPN, who are some distance away from where they live.
“They’re still under the care of their local haematologist, but they can have a second opinion or more direction if they’ve got tricky management problems with someone more experienced in MPN, without taking a day off work, without travelling, and certainly without crossing borders, which has been completely impossible.
“Some patients previously went to great lengths to travel to Gosford, where I am, and being able to offer telehealth is very time efficient for patients.
“I see a lot of patients with MPN, including second opinions, and have a little group of patients who’ve travelled to see me and whom I share care with their local haematologist,” explained Dr Forsyth. “I actually have a patient who has travelled from Far North Queensland for a second opinon.”
“He came down to Sydney for a reason, a couple of years ago, and saw me to ask me about interferon. It was in the early days of interferon being funded by the Pharmaceutical Benefits Scheme (PBS) for myeloproliferative neoplasms. His local haematologist hadn’t had much experience [with it].
“I still catch up with him once a year, which is much easier now there’s telehealth. I can have a good chat to him, and he’s shown me around his house and introduced me to his pets.
“It’s really a very enjoyable way of consulting,” said Dr Forsyth.
“I never have any hesitation, for my own patients, about organising a second opinion for them.
“And I always feel that if a patient wants a second opinion I try and ensure that it is opinion and advice that will benefit the patient and that maybe I would learn from also,” said Dr Forsyth, who is happy to make the referral.
“I tailor the recommendation to a person who is appropriate for the patient to see, and that’s not necessarily their next-door neighbour’s brother’s cousin’s uncle!
“If I organise the referral then patients can take all their results with them, a copy of all the pathology, all the imaging, and I can write a letter about why I’ve chosen what I recommended.
“I say to patients… if you do want to seek a second opinion, raise this with your treating haematologist. Talk to them about it and, if you feel comfortable, tell them why you would like a second opinion.
“Some haematologists don’t like it and feel upset about it. But we have to remember this is not about haematologists’ egos, it’s all about the patient and their disease, and whatever is best for the patient is fine by me.
“Sometimes people seek a lot of different opinions. There are patients that I have been the fourth haematologist they’re involved with, and I think that gets challenging for the patient.
Sometimes patients seek a second opinion because they haven’t got a good working relationship with their first haematologist, and you do need that.
“When you have a chronic illness, you need to be able to communicate effectively and feel that your haematologist cares for you and tries to assist you as best they can with your management.
“In the global era, sometimes people get advice over the internet in support groups that comes from outside Australia, where therapies are suggested that aren’t available or funded here.
“Patients need to bring these kinds of things up with their treating haematologist and say, I would like a second opinion. Sometimes they can phrase it as their family is keen for them to get a second opinion. It can be a bit easier to raise it from that perspective,” said Dr Forsyth.
Dr Forsyth encourages her patients to consider a wide range of physical activities. Here she is kayaking in Norway.
How Dr Forsyth got into medicine, haematology and MPN
“I didn’t come from a medical background but was always interested in medicine and liked people, and being interested in science, the idea of going into medicine appealed.
“I enjoyed each term I did as a junior doctor, from intern to resident, and thought, ‘oh, I could be a gastroenterologist, or I could be… whatever.”
“Then, when I did haematology, I just loved it.
“I liked looking after people who were significantly unwell; that didn’t seem daunting. I didn’t mind talking to people about their life-threatening illnesses, and I liked the ability to do pathology at the same time; to see what their bone marrow looked like under the microscope, and care for the same patient,” said Dr Forsyth.
Today, due to her clinical load, Dr Forsyth is “just a clinical haematologist” but one who finds MPNs “an interesting group of illnesses”. This intensified “about two decades ago” when she took over the Australasian Leukaemia & Lymphoma Group’s PT-1 study as principal investigator from Professor Andrew Grigg.
“From there my passion for MPN has grown,” said Dr Forsyth and she calls herself “a community haematologist”.
She sees whatever type of haematology walks in the door of her practice on the Central Coast of NSW, 100km north of Sydney, but she has a love for MPN, which she describes as “a chronic blood cancer resulting in the overproduction of mature blood cells and associated with a wide range of significant symptoms that impact on quality of life”.
Access to MPN treatments
“We can do things better than we do for MPNs and that inspires me to raise patients’ awareness that there have been advances in diagnosis and management,” said Dr Forsyth.
“Overall, as a group, people with MPN are getting good care in terms of what is available for those patients.
“But sometimes you may have a haematologist who’s not experienced in using interferon [peginterferon alfa-2a (Pegasys®)].
“There has not been much pharma support available for haematologists and patients because of the very strange way it was PBS-funded, without a TGA indication,” Dr Forsyth explained.
“That’s why the MPN Alliance patient information sheets on interferon are so critically important in providing information for patients, and the clinicians about how to use interferon.”
The PBS-funding of Pegasys for patients with MPN, in 2018, “was really fantastic”, Dr Forsyth said.
“We’re still rejoicing in having pegylated interferon, as opposed to non-pegylated interferon. It’s been a good opportunity for patients.
“I think we will continue to see more and more data about the potential for interferon to modify the natural history of MPNs.
“Internationally, there is increasing evidence of the role of interferon and there’s an ultra-long-acting interferon which we don’t have in Australia, called ropeginterferon alfa-2b, that has less toxicity and equal efficacy [as Pegasys].”
Dr Forsyth, hiking in Ethiopia with her husband, James Rogers, son, Harry Rogers, and daughter-in-law, Subeta Vimalarajah
Dr Forsyth said, disappointingly, ruxolitinib was not available through the PBS for Australian patients with PV who can’t tolerate or who have failed treatment with hydroxycarbamide (formerly known as hydroxyurea).
“They can’t get ruxolitinib until they’ve transformed to myelofibrosis (MF).”
Dr Forsyth said many centres are participating in clinical trials looking at adding other drugs to ruxolitinib for patients who are newly diagnosed.
“And for patients who have failed ruxolitinib, or are losing their response, studies are looking at what other drugs can be used to improve the response rate in that group.”
Dr Forsyth said the outcomes for young people–diagnosed under the age of 50 years–with ET was very good; they have a near normal life expectancy.
“Most people with ET and PV have a long survival, but on the other end of the spectrum–patients with MF–some of those with very high-risk disease have very short survival.
“Patients with high-risk myelofibrosis at diagnosis, are rapidly moved to bone marrow transplantation as their only curative option,” said Dr Forsyth.
And the group of patients with higher risk MF who are not eligible for transplant want to be participating in clinical trials, she said, “to try and improve their outcome because, unfortunately, MF can be a rapidly progressive and incurable malignancy”.
“There are more studies available with drugs that are more exciting than any other time before, and that’s really encouraging.”
“There’s very good data to show that patients who participate in a clinical trial do well and have better outcomes. That’s because they have access to treatments they wouldn’t have otherwise.
“Some patients may be motivated to participate in a trial because they are contributing to research that may help them and certainly may help other patients down the track,” said Dr Forsyth.
“I like to refer patients to the haematologist who is running a clinical trial, to make sure they have all the information they need and to minimise tests being repeated.
Information on clinical trials is available from ClinTrial Refer
Unmet needs in the MPNs
Dr Forsyth said all the MPNs can transform to secondary AML, many of them by transforming first to MF.
“This is a very unmet need, and AML that has arisen from MPN has a very poor outlook and, in that circumstance, can be very challenging.
“Often patients who you’ve known for a long time with their ET and PV, can suddenly develop changes that are consistent with transforming to AML. It can be very distressing for the clinician as well as for the patient,” she said.
“When transformation to AML occurs it is important to carefully consider what you can offer the patients, how successful treatment is likely to be, and their quality of life.
“For many elderly patients who transform to AML, we choose not to give them induction chemotherapy but offer them supportive and palliative care because, unfortunately, the outlook is grim.
“I think patients have to be realistic,” said Dr Forsyth. “Do you want to spend that time in hospital, having treatment that is unlikely to prolong your life, or do you want to be at home? Those kinds of discussions are difficult for everybody, most importantly, for the patient receiving that news.
“I try to reassure patients that the likelihood of transforming to AML is low.”
“I tell them there is a risk and that usually occurs when the disease has been present for a long time, but that risk is low, that risk is down the track, and let’s look at optimising what we can do.
“Unfortunately, there is no definite evidence that we can alter the natural history of the disease with currently available therapy.
“There is single centre evidence that interferon may have a role, so I might discuss that with patients when considering what cytoreductive therapy to recommend.
“Unfortunately, it does take patients time to cope following the diagnosis of an MPN especially when they realise that it is a blood cancer which is generally incurable.
“For many people, it will never progress, but it really is important in whatever circumstances to live your best life. I think that worrying about tomorrow today is very problematic and I try and move patients on from doing that too much.
“Worrying about what might happen tomorrow can really detract from how you live today.”
“A minority of people are so worried and consumed with anxiety about when their disease may or may not progress that they can’t enjoy actually being well today.
“We need to help those patients and I refer them to an oncology psychologist.”
Advice for the newly diagnosed
Getting advice and information from reputable sources is Dr Forsyth’s key tip.
“I encourage them to live their best life, tell them there are things they can do to help with their life and that we can manage and control their disease to ensure their quality of life is as good as it can be. I tell them they can live a normal life, hopefully for a very long time, and in some cases with a normal lifespan.
“I try and give them a message of optimism, of self-control of symptoms, and motivate them about what they can do that helps with their quality of life.
“And I try and encourage them to get good support and use good sources of information from the internet and warn them that there are some less reputable sources of information on the web that should be avoided.”
*Professor Ruben Mesa is an international MPN expert who is committed to improving therapeutic options and quality of life for people with MPN, as well as being an active patient advocate.
ET or PV? That’s the question Sue’s been pondering for years
Since her diagnosis with essential thrombocythaemia (ET) in 2013, Sue Taylor has been curious about whether she may in fact have a different type of MPN–polycythaemia vera (PV).
“It certainly was always at the back of my mind,” said Sue, 64, of Sydney.
Sue Taylor
Her search for the answer has seen her change her treating haematologist several times and go overseas to an MPN conference, so she could put the question to international experts.
Sue’s main interest in having an accurate diagnosis centred around her risk of thrombosis (blood clot) but was also about possible future access to new treatments and clinical trials
In 2009, soon after Sue took a voluntary redundancy from her sedentary but stressful job in the Australian Public Service, she got “some strange visual symptoms” which “was a bit concerning”.
When an ophthalmologist couldn’t find anything wrong, Sue put these episodes down to “just something happening” and got on with life, despite increasing episodes. (Later, Sue found out she had scintillating scotoma)
Sue and her husband, John, have spent much of their lives together working overseas and travelling extensively, especially after her retirement.
“We’ve packed a lot into our life, and it has been wonderful,” said Sue.
“We were in a fortunate position to be able to take a bit of time for us, while fit and healthy, to enjoy some time together doing things we wanted to do. For us that meant a lot of travelling.
“Having three close family members pass away in the recent past made us think you can’t take anything for granted.”
Four years on, when Sue went to her GP, a routine blood test showed her platelets were “fairly elevated” and, looking back at previous years’ blood results, it was apparent that although in the normal range, her platelet count had been increasing.
“At this point I was sent off to a haematologist and had the JAK2 test. It was positive and I was diagnosed with ET.”
“I felt very well and would never have thought I had anything wrong with me,” said Sue, then aged 53.
“Honestly, it’s a shock when you’re first diagnosed, but I was reasonably philosophical and just grateful it wasn’t something worse. I’d recently seen my dad go through motor neurone disease.”
Sue with her grandson Teddy when he was six months old
Her haematologist didn’t seem to think her visual symptoms were significant but put her on baby aspirin due to her high platelet count.
“The amazing thing, which I didn’t realise until a year later, was… I never had another of those episodes… except once, when I went off aspirin before having surgery.”
Being “an extremely curious person” and studying genetics for her science degree at uni, Sue had “done a bit of reading” on the internet about MPN before seeing her haematologist.
“I was told there was nothing to worry about but if my platelets continued above 600, then he might think about putting me onto hydroxyurea.
“When I asked about a bone marrow biopsy he said, ‘you don’t need to know about that yet’, which was like a red rag to a bull. He really wasn’t the right haematologist for me.
“I’m a very research and evidence-based sort of person, so I tried to see someone who was into research and preferably had an interest in MPN.
“I was fortunate to be linked up to a really special haematologist, who was involved in research, but MPN wasn’t his thing,” said Sue.
“Then I read about this third person who had an interest in PV and had been involved with PV study groups earlier in his career.
“I was referred to him, which was alright until he retired.
“He’d written a paper on platelet-mediated visual disturbances and was the first person who actually thought my visual disturbances might have some relationship to my ET. He was a bit excited because he thought I was a classic case.
“In the meantime, I did more reading and learnt a lot. I discovered MPNs are quite rare, there wasn’t a lot of research being done, certainly not in Sydney that I could find,” said Sue.
She came across an Italian research paper about JAK2 and PV.
“They worked out that a haematocrit–a measure of the volume of red cells in your blood–of 45 or below was the range for people with PV, to avoid a thrombosis which is the main immediate concern of haematologists treating MPNs.
“Although I’d been diagnosed with ET, I was very curious about this because my haematocrit was regularly measured at 47-48. I was concerned I was at risk of thrombosis, having a haematocrit that high.
“The JAK2 mutation was only discovered in 2005 and there were debates and different points of view about whether ET and PV were different diseases.
“There’s been research which shows a lot of people have been incorrectly diagnosed with ET who actually have PV,” explained Sue.
“I wondered if I had ‘masked PV’; a type of PV where you don’t quite fit the criteria to be diagnosed with PV, in terms of your blood counts, but it can be masked because of very low iron levels in your blood.
“I had very low iron levels, but I really couldn’t get any haematologist to be terribly interested in that. They were always interested in my platelets, but not my haematocrit.”
Sue Taylor arriving home after picking up her first packet of Pegasys from the hospital in August 2018. “It’s all packed up in ice!”
In 2017 Sue went to a patient conference, held by the MPN Education Foundation every two years in the U.S., that brings together eminent MPN researchers and clinicians from around the world.
“It was a fantastic experience. I met some of the MPN patient representatives in the U.S., and other patients.
“I still didn’t really get my question answered but I was told that, to be on the safe side, I should make sure my haematocrit was lower.”
Sue heard about pegylated interferon alfa-2a (Pegasys®) at the conference, and that in some cases it can lead to molecular remission.
“In Australia, haematologists were only talking about hydroxyurea, which I was concerned about taking because it exacerbated non-melanoma skin cancers, and I have very fair skin.
Sue had been on watch and wait and her only treatment was aspirin, until 2016, when she started having “funny spells”, didn’t feel very well, and her blood pressure was “a bit up and down”.
“I agreed reluctantly to go on hydroxyurea, just to get my blood counts down and to see if that made any difference,” said Sue.
“My understanding was that hydroxyurea could be quite good for controlling your blood counts, but it didn’t do anything about controlling possible disease progression to two other versions of MPN–very active PV and myelofibrosis (MF).
“One thing MPN patients are very interested in is the risk of disease progression and how they might minimise that.”
“It’s only a low risk but it’s there, and there’s really no way of predicting who will progress. There’s still a lot that’s unknown.
“I was always very interested in interferon but any haematologists I spoke to in Australia at that time said the side-effects were difficult, and ‘you don’t want to be on it’.
“I was aware pegylated interferon was available in Australia on compassionate grounds, and knew efforts were being made to try and get it on to the Pharmaceutical Benefits Scheme (PBS).
“At the time it was about $1400 a month… more than I wanted to pay.
“I was really grateful when Nathalie Cook (a MPN patient and advocate) was successful in her effort to get Pegasys on the PBS and I thought, ‘yes, I’d really like to give it a go’.
“As soon as it became available, I asked my haematologist to put me on it and I had to be assertive in my request. I was her first patient on it for MPN,” said Sue who had been on hydroxyurea for 18 months.
“Once I went on Pegasys, I was extremely relieved and surprised. I don’t think I’ve had any side effects and I haven’t had any headaches,” said Sue.
“It takes a bit of getting used to, injecting yourself, and I got advice and tips from other patients that was very helpful.
“I was able to live my normal life. It really didn’t impact me. And I feel incredibly grateful for that.
“In my case, it worked very effectively at a very small dose and I’ve actually been able to halve the dose. So, I only use it every fortnight and I’m trucking along quite well.
Sue with her husband, John, on their travels
“My bloods are all within normal range.
“I don’t want to sound as though I’m an advocate for it, because everybody’s got their own response to different treatments, but for me, it’s worked well so far.”
Sue’s been on Pegasys for more than two years. This includes a four-month break when she “had an episode” and ended up in hospital with a high fever and severe chest pain.
“We’re not sure if that was related to the Pegasys or not, but I’ve been back on it again for a year and it hasn’t happened again.
“In the course of that, I sought another opinion from a haematologist with more experience with interferon, and other medical specialists.
“I was very well looked after, and my current haematologist was very good at facilitating all that too.”
“And that haematologist, who I saw for a second opinion, looked over my full history and was suspicious that I may well have masked PV, which was interesting.
“I still have the diagnosis of ET formally but I’m not so worried about it now, so long as I’m on a drug that is keeping my haematocrit under control.”
“My haematocrit has been below 45 since I’ve been on treatment so at least I feel comfortable that any thrombosis risk is being managed.
“The other reason I was really keen to have an accurate diagnosis was that when new drugs come along or clinical trials, they usually have pretty specific criteria as to who can have them,” said Sue.
“Who knows what might come along in the future.
“There are some really positive results coming out of clinical trials into a new type of interferon alfa-2b, called ropeginterferon (Besremi®). It’s a slightly different formulation and is longer acting.
“It’s been tested on people with PV, and the results have been quite good as reported at ASH [the American Society of Hematology annual meeting] and in Europe at some haematological conferences.
Sue Taylor when she attended the MPN patient conference in Scottsdale Arizona: “the Mayo Clinic is in the background”
Sue feels very fortunate her MPN hasn’t held her back in life “at all”. Her response to this “strange diagnosis” was to focus on having a healthy life and doing as well as she could by eating healthy food and controlling her weight.
She was already doing pilates and bushwalking, so she joined a gym and went several times a week for her cardio fitness.
“The joy of using your body and feeling strong and fit I found to be an unexpected benefit… a buzz. I felt really good,” said Sue, but she developed an ankle problem which has taken “some years” to get better, with “months and months” of rest.
“I’ve got severe arthritis in my ankle. That’s probably the worst thing that’s happened to me.
“It’s the inability to do what I love most, which is being physically active.
“I still do pilates and walk, but only on the flat,” said Sue who is careful with her diet.
“I figured that if you’ve got an ankle injury or knees or hips, keeping your weight at a good level is a good thing, and it also helps with your mental wellbeing.
“I eat more of the good stuff and less of the not-so-good stuff” said Sue.
That means more good carbs, vegetables and fruit, fish, small portions of meat, more beans, nuts and seeds, and not eating between meals. She still treats herself to a slice of birthday cake and dark chocolate, going for quality not quantity.
Sue Taylor, when she went to the MPN patient conference in 2017
Sue tends to think of MPN “as a chronic condition, like diabetes”.
“I’m focusing on doing what I can to maintain my general health and fitness.”
“People can be worried by MPNs being called ‘cancer’, which has the unfortunate effect of frightening people. I can understand why it’s called a cancer, but there are benefits for MPNs to be cancer, including cancer funding and getting more notice from doctors and researchers.
“I have never felt frightened of my diagnosis. My intellectual curiosity to find out more about MPNs is my way of taking control.
“I was given no information,” said Sue about when she was diagnosed.
“When I moved to my second haematologist and was in the waiting room of a big teaching hospital in Sydney, I found all the Leukaemia Foundation booklets. I was very excited to see the MPN one amongst the others. It gave me some basic information.”
Travelling is not so high on Sue’s agenda these days. She’s providing support to her elderly mother and has two grandchildren to enjoy time with, and one on the way.
“We’re a pretty close family, so we like to see as much of them as we can,” she said.
Looking back on her journey with MPN, Sue has realised it “takes time to adjust”. From her interaction with other MPN patients, she understands that everyone has a different experience with their MPN, and there is no “one size fits all.”
AL amyloidosis trials due to open this year offer “new hope”
Dr Simon Gibbs at an international haematology conference in Boston, in 2019
Two clinical trials will open later this year for newly diagnosed patients with AL amyloidosis with moderate to severe cardiac disease.
These studies will hopefully address a major unmet need, according to Dr Simon Gibbs, Director of the Victorian and Tasmanian Amyloidosis Service, and a founding member of the Australian Amyloidosis Network (AAN).
“The big challenge we have is that group of patients who have been diagnosed late with severe cardiac disease,” said Dr Gibbs.
“We know that, unfortunately, up to a quarter of these patients still die within six months of diagnosis.”
Dr Gibbs said the other 75% “often enjoy really good responses to treatment” and “we know that patients have very good outcomes if we get control the excess production of light chains quickly”.
“Not everyone responds as well as we’d like to our standard upfront therapy, VCD (Velcade®, cyclophosphamide and dexamethasone),” he said.
“Daratumumab is clearly an effective therapy to rescue patients who have poor responses to VCD, and the ANDROMEDA study proved that using daratumumab with VCD improves response rates.
“But we really need more rapidly effective treatments for patients presenting with advanced cardiac disease, or who don’t respond well enough to VCD.”
“We are hopeful two upcoming trials, the AFFIRM-AL (birtamimab) study and the CAEL-101 trial, will help address this unmet need, help to overcome early mortality, and may offer advances in patient quality of life and survival,” said Dr Gibbs.
Both Phase III global trials are looking at the standard therapy for AL–VCD–plus either birtamimab (previously called NEOD001) or CAEL-101. These drugs are monoclonal antibodies that target the amyloid deposits, and an aim of these studies is to clear the amyloid from affected organs.
Birtamimab was studied previously in the VITAL study. While the overall patient group in this trial did not significantly benefit with the addition of birtamimab to VCD, there was a strong suggestion that patients with advanced cardiac disease obtained the most benefit, and may have enjoyed better outcomes than the group that received VCD alone.
“One problem with the VITAL study was that there weren’t enough patients in the advanced cardiac group to be sure that birtamimab was beneficial, but the signs are good. So we are hopeful the AFFIRM-AL study will show a positive result,” said Dr Gibbs.
These two trials will look solely at people with advanced cardiac disease at diagnosis, to see if these drugs, which help to strip away the amyloid, have benefits for this group.
“We are always on the lookout to ensure our patients connect to the world’s best novel treatments and clinical trials,” said Dr Gibbs.
“The good news is that patient outcomes are improving overall.
“We’re finding that patients with mild to moderate disease at diagnosis generally do very well and tend to be living a lot longer.”
“Most patients will obtain a good remission from their AL amyloidosis.
“Patients are surviving much longer than 10-20 years ago,” said Dr Gibbs.
“This is because we now have access to newer, more active drugs like bortezomib (Velcade®), and lenalidomide (Revlimid®).
“We are also much better these days at choosing which patients will benefit from stem cell transplants, and daratumumab has recently been shown to be of significant benefit to many patients with AL amyloidosis, which is a very exciting advance,” he explained.
“In addition, there’s a greater understanding of medications that can help support the affected organs, and there are more amyloidosis centres of excellence around the world, so there’s more knowledge.”
Another trial will soon be available for relapsed AL amyloidosis
Dr Gibbs said a third trial, for relapsed/refractory AL amyloidosis, also “will hopefully be starting at the end of the year if all goes well”.
“It’s an Australian-French collaboration, which we are very excited about.”
Expert Series: Dr Simon Gibbs on all things amyloidosis
Dr Gibbs at Gard du Nord Paris
Dr Simon Gibbs is a consultant haematologist (Myeloma/Amyloidosis Lead) at the Department of Clinical Haematology Eastern Health (Melbourne), Director of the Victorian and Tasmanian Amyloidosis Service, a founding member of the Australian Amyloidosis Network (AAN), and Honorary Senior Lecturer at Monash University. In this far-reaching interview, he discusses everything from the incidence, diagnosis and treatment of amyloidosis, to supportive care, quality of life, and how Dr Gibbs came to specialise in this field.
What is amyloidosis and how common is it?
Dr Gibbs describes amyloidosis as “a group of diseases where proteins within the body kink or misfold, then stick together abnormally and deposit in organs causing them damage. Once enough of these sticky misfolded proteins or amyloid ‘fibrils’ deposit in the organs, they can cause the organs to fail”.
“Amyloidosis is not as rare as we once thought it was. There are many types, but the two most common are AL and TTR,” said Dr Gibbs.
“In AL, the misfolded proteins are excessive amounts of light chains, which are part of the immune system and produced by the bone marrow.
“In TTR, the misfolded proteins are transthyretin, a protein mostly produced in the liver, but the amyloid fibrils mainly affect the heart.
“Unfortunately, both TTR and AL amyloidosis are under-diagnosed or diagnosed late. We know many patients will see several specialists over many months before the diagnosis is made.”
“We still have the challenge that amyloidosis can be difficult to diagnose.”
“Fortunately, in the last decade we’ve discovered there’s an easier way to diagnose TTR amyloidosis, by using a special bone scan. This has led to greater awareness of this disease, so patients are being diagnosed and treated earlier, and hopefully that equates to better quality of life and overall survival.”
Dr Simon Gibbs on the Hong Kong Star Ferry.
And there’s more awareness of AL amyloidosis now than six years ago, when Dr Gibbs returned to Australia from the UK.
“AL amyloidosis is a disease of adults. We don’t see it in children, it’s unusual before the age of 45, and the incidence seems to peak in the mid-60s.”
According to a Queensland study, approximately 12 people per million are diagnosed with AL amyloidosis, and there is a slight male predominance.
In TTR amyloidosis, 85-90% of patients have wild type (or non-hereditary disease) and 90% of these cases occur in men, with the average age at diagnosis being 75-80 years.
In the 10-15% of people with of TTR that is hereditary, the disease affects men and women equally and is more likely to involve the nerves. This can cause numbness, pain or electric shock-like feelings in the feet, cramping of the legs, weight loss, bowel disturbance, and dizziness.
Dr Gibbs said TTR amyloidosis appears to be a disorder of ageing. In Japanese and Finnish studies that looked at autopsies of people aged 90 or older when they passed away, it was discovered that 25% of these people had amyloid deposited in their hearts.
“It seems that if we live long enough, we’ll probably all develop TTR amyloidosis!”
“We think there are about 7000 people with TTR amyloidosis in Australia, but this is a wild guess.
“Detection of this disease is definitely increasing. In 2016, we had one new referral a month to the Victorian and Tasmanian Amyloidosis Service, now we’re receiving one or two per week.
“In Melbourne at least, this disease is seen mainly in Caucasians, particularly of Italian heritage, and we don’t see it so much in people of Asian backgrounds.
“Interestingly, there seems to be a slight predominance in people who have been elite athletes in their youth,” explained Dr Gibbs.
“I have a number of ex-AFL footballers who have TTR amyloidosis.”
Dr Gibbs said a ‘red flag’, or warning sign, for TTR amyloidosis is any man aged over 60 who has had both his carpel tunnels released.
“So, if a man has had a carpel tunnel release operation on both wrists, our rule of thumb is that you should always consider that they are at high risk of developing TTR amyloidosis. If these men present with shortness of breath, unexplained weight loss, fatigue, or swollen ankles, they have TTR amyloidosis until proven otherwise, and they should immediately have a bone scan.”
“That’s one of the messages we are really trying to deliver now to cardiologists.”
One of the differences with TTR amyloidosis, compared to AL (which can affect almost any organ), is that typically TTR only affects the carpal tunnels, the heart, and spinal ligaments.
“Over 70% of TTR has had carpal tunnel syndrome, which usually develops 10-15 years before heart problems emerge. Many, especially ex-sportsmen, will also have had laminectomies or spinal surgeries in the past.”
At the AAN Amyloidosis 2019 meeting, Simon Gibbs, second left, with Dr Fiona Kwok, Professor Angela Dispenzieri and Associate Professor Peter Mollee
Dr Gibbs wonders whether tissues in the body that have been used a lot are more prone to laying down TTR amyloid fibrils, such as the hands, back, and heart, and “sportspeople tend to use these a lot, but we really don’t know”.
“There are lots of unanswered questions and that’s one of them.”
But Dr Gibbs said the good news is that, over the last five to 10 years, the availability of medications that can slow progression of TTR amyloidosis has increased.
“The advantages of attending an AAN centre is access to drugs that you can’t get elsewhere, such as diflunisal, which, if tolerated is a good treatment for TTR, especially neuropathic disease; compassionate access programs for drugs like tafamidis; or clinical trials of newer therapies, such as patisiran or inotersen.”
And Dr Gibbs said trials of gene-silencing medications to reduce the production of TTR look particularly promising.
With other non-TTR hereditary forms of amyloidosis, Dr Gibbs expressed his frustration that there still were no specific treatments for those patients.
“Sometimes we put our patients on doxycycline – an old antibiotic – which may slow the misfolded proteins sticking together, so hopefully slowing the disease a little bit, but a lot more research in this area is desperately needed.”
Dr Gibbs highlighted the point that the amyloid proteins in Alzheimer’s disease are “completely different” to the amyloid proteins seen in AL or TTR amyloidosis, “so the risk of developing Alzheimer’s for someone with TTR or AL is the same as everyone else [in the community]”.
Dr Gibbs, centre front, at a Medical & Scientific Advisory Group meeting
How Dr Gibbs got into medicine, haematology… and amyloidosis
While at school, Dr Gibbs aspired to being in a profession “where you help people” but when his parents worried that medicine would be too much work, he considered being a lawyer or an accountant.
“I did work experience in a law firm but ran away from there thinking, “Oh my God, there’s no way I could do law!”.
Dr Gibbs didn’t think he was clever enough for medicine until he received his final school results.
“I started medical school and felt incredibly dumb because I went from being one of the bright kids in the classroom at school to being a very average student at university,” explained Dr Gibbs, but he persevered through the six years of medical school.
He trained at Melbourne University, his internship was at the Royal Melbourne Hospital, then he went across to St Vincent’s Hospital as a second-year junior doctor where his first rotation was in haematology.
“Medicine, as something to study, is very, very interesting and once I started seeing patients, it became a lot more rewarding.
“I worked under some amazing doctors. I was the junior resident to Andrew Wei, who was the haematology registrar at the time.
“Andrew has gone on to become Professor Wei, and is Australia’s leading expert in acute leukaemia.
“Andrew is just such a brilliant, brilliant doctor and mentor. He inspired me, so I decided to enrol as an advanced trainee in haematology at the Peter MacCallum Cancer Centre and St Vincent’s.
“I really enjoyed haematology for the fact that you really get to know the patient and you go on a journey with them, supporting them from diagnosis through often quite a few months of treatment, then hopefully remission and even cure.
“It is a very rewarding speciality, in my opinion.
“Haematology has always excited me because it’s a fast-moving field. It’s evolving. There’s a lot of research going on, so I always found it very interesting, and I’m a naturally inquisitive person.”
Dr Gibbs, out and about in Nice while attending a meeting there.
As a junior haematology registrar, Dr Gibbs had a patient with AL amyloidosis.
“It was pretty clear to me that no one really knew back then how to best treat what was thought to be a very rare disease, and I had a patient die a pretty horrible death on me, and I thought, ‘Oh, God. That’s awful’.”
Then Dr Gibbs had an opportunity to complete his final year of haematology training in Cambridge in the UK. His boss at the time, Professor Nandurkar, now Director of Haematology at the Alfred Hospital, suggested that he return to Australia from the UK with a ‘niche’ or new skill not readily available in Australia. At Cambridge, Dr Gibbs saw more amyloidosis patients.
“I heard about this National Amyloidosis Centre [NAC] in London. They compiled these terrific reports on patients, very informative, and I thought, maybe that’s my niche,” said Dr Gibbs.
He went down to London and met with the Director of the NAC, Professor Philip Hawkins, and was offered a job on the spot.
“He said, ‘not a lot of people are interested in amyloidosis’.”
Dr Gibbs enjoyed his two-year amyloidosis fellowship so much, he stayed on at the NAC for an additional 2½ years! Dr Gibbs admits that, not only were the doctors he worked with there great mentors, but the opportunities in London, and the ability to travel regularly to amyloidosis meetings all over the world, made him stay on longer than originally planned.
He then moved to northern England, to lead the huge myeloma service at the Manchester Royal Infirmary for more than two years. While there, he set up the first satellite clinic for the NAC.
Dr Simon Gibbs walking in Scotland
Returning home and setting up Australia’s third Amyloidosis Service
Dr Gibbs wasn’t sure he could “put up with English winters” for the rest of his life, so when he was approached by Victoria’s Eastern Health to help establish their new myeloma service, he jumped at the opportunity to return home. But he ensured that the job allowed for setting up a dedicated amyloidosis service – the first in Melbourne.
Dr Gibbs returned to Australia in 2014 and founded the Victorian and Tasmanian Amyloidosis Service in close liaison with the two existing amyloidosis clinics in Australia, at the Princess Alexandra Hospital (Brisbane) and Westmead Hospital (Sydney).
“In 2016, our three services met and agreed not to compete but to collaborate, so we set up the Australian Amyloidosis Network,” he said.
“The idea was that we would join forces to help enhance awareness of amyloidosis, services and supports to patients, and to attract new treatments and clinical trials to Australia.
“At the time, Australia was being overlooked for clinical trials in amyloidosis.”
“We were delighted when, in 2018, the Fiona Stanley Hospital in Perth agreed to join the AAN, under the leadership of Dr Hasib Sidiqi who’d just returned from a fellowship in amyloidosis at the prestigious Mayo Clinic in the U.S.
“Since Hasib joined, I think we’ve been pretty successful in bringing important international clinical trials to Australia. Fortunately, we have some terrific colleagues in Adelaide also.”
Since forming the AAN, the two leading Prothena studies in AL amyloidosis – VITAL and PRONTO – as well as Janssen’s ANDROMEDA study, the largest ever clinical trial globally in AL amyloidosis, were offered to Australian amyloidosis centres.
“With the exception of Greece, Australia had more patients per capita than any other country enrolled on the ANDROMEDA study, and it meant we could give daratumumab to some our patients that we otherwise couldn’t do.
“Then the Eidos AG10 study was our first clinical trial in TTR amyloidosis. This drug looks very promising, and we await the initial results of this study, which hopefully will be released in early 2023.
“We now have four really good TTR trials running in Australia” which, Dr Gibbs said, “is just amazing”.
Details of these trials are available on the AAN’s website.
“I think supportive care can be just as important in many cases as the chemotherapy itself,” said Dr Gibbs.
“One of the pieces of advice I give patients is… if you’re getting a symptom that’s really upsetting or bothering you, really jump up and down about it, because we do have treatments that can help most symptoms.
“A haematologist is very good at giving chemotherapy, but we know that AL amyloidosis patients have more complex needs than myeloma patients because of their organ involvement.
“I suggest to patients, many of whom have three or four specialists… always ask all your doctors to copy each other in on letters and reports, and always bring a copy of your medications to your consultation.
“An amyloid specialist acts like a conductor of an orchestra. We will often be the central person for the cardiologist, the local haematologist, the kidney specialist, and the neurologist to communicate with about the coordination of the patient’s care.
“Sometimes we doctors aren’t always good at communicating regularly and effectively with each other, and so we might not necessarily know of changes to medication or results of scans ordered by other doctors.”
Quality of life advice
Dr Gibbs said, for a lot of patients with AL amyloidosis, poor appetite is a major problem.
“Normally, when we start eating, the stomach relaxes so it can fill up with all the food from a full meal. In amyloidosis, this reflex is lost or is sluggish. So when the stomach doesn’t relax, patients feel full more quickly.
“We call this ‘early satiety’ or ‘gastroparesis’ (paralysis of the stomach).
“Patients usually have a good breakfast, barely anything for lunch, and skip dinner because half their breakfast is still sitting in their stomach by the time dinner comes around.
“One of the easiest things to do for AL patients who say, ‘I’ve lost a lot of weight’, ‘I’ve no appetite’ is put them on a ‘prokinetic’ drug. These medications help push food through the stomach, so patients can eat a bit more.
“Medications such as domperidone (not Dom Pérignon!) or metaclopramide can really help improve patients’ appetites and maintain their weight,” said Dr Gibbs, and he puts at least half of his patients on domperidone.
“And all of a sudden, patients can go from only having two small meals a day to maybe one full meal and another two half meals. They eat a bit more, meaning they have a bit more energy, less chronic fatigue, and they stop losing weight. This can be a big psychological boost for patients, as well as a physical one.
“It’s such a simple thing, starting domperidone. It’s very helpful and can make a big difference to quality of life.”
If you are losing weight, Dr Gibbs said starting dietary supplements like Sustagen, and seeing a dietitian, can be helpful.
“If you’re struggling to maintain a good weight, whatever you’re eating, make sure it’s full of lots of calories.
“Try to eat things that are high in protein and calories that are easy to digest. Minced meat is easier to digest than a steak, so things such as cottage pies and Bolognese sauces can be good. They are easier to swallow, easier to pass down through to the stomach, and easier to digest.
“Try drinking cordial rather than water, or maybe have a soup or protein shake, and add a spoonful of honey to your tea.
“And try to avoid of lot of bread. Many people say bread is quite difficult to digest. It sits in the stomach for a long time and doesn’t move through easily.”
Professor Philip Hawkins, centre, with Simon Gibbs and National Amyloidosis Centre Fellows in the UK.
The ALIGN study
Nutrition is a poorly studied area in amyloidosis, according to Dr Gibbs, and the Victorian and Tasmanian Amyloidosis Service has begun a clinical study, called the ALIGN study, for newly diagnosed patients.
It’s looking at their dietary history, liver size, and the transit time of food through the stomach (through gastric emptying studies).
“We’re collecting information about weight, diet, and conducting quality of life and supportive care screens.
“The idea is to really look at diet and quality of life and the use of prokinetics, such as domperidone, and see how much of a difference they make and who benefits the most. Anecdotally, I think it makes a huge difference, but we are in the process of looking at this more scientifically.
“We want to conduct these investigations, if possible, before treatment starts.”
As it is still early days regarding results, Amyloidosis News will report back on the findings of this ongoing study in two years.
Dealing with neuropathy
Peripheral neuropathy can be caused by the amyloidosis itself or, in some circumstances, from treatment, such as with bortezomib (Velcade®).
“Once intermittent neuropathy symptoms became more regular, we need to question, ‘what are your light chains and have we achieved a good enough response?’ so we can decrease the dosage of bortezomib to control these symptoms or even consider a treatment break,” said Dr Gibbs.
“Often we’ll give six cycles or roughly six months of treatment. If you obtain a 90% response within the first cycle or two, but you’re starting to develop neuropathy by month three or four, we can probably reduce your bortezomib by half or space it out to fortnightly dosing, if you’ve got to where you need to be.
“There’s no point suffering from constant leg pain or dizziness,” he said.
“Various therapies can help treat peripheral neuropathy symptoms such as electric shock feelings or pain in the legs.
“Lyrica® and Endep® are medications that help to decrease the sensitivity of the nerve endings. They can cause a bit of drowsiness, so most patients take these at night before going to bed, which helps with sleep.
“And, start on a low dose and slowly build up to levels that are going to be helpful. The other thing is to be patient! These treatments can take up to six weeks to kick in and work.
“When starting any new medication, think about taking the first few doses at night, because if you develop side-effects, hopefully you’ll be asleep when you get them, so they won’t feel as bad. Then your body starts to get used to the medication,” said Dr Gibbs.
“And for general pain, two tablets of good old Panadol will decrease the intensity, but you have to take it regularly, don’t wait for the pain to hit. If you take it when the pain has already started, it’s not going to work.
“If patients develop cramps, magnesium, and quinine, which is found in tonic water, are quite good.
“For dizziness when you stand up, sometimes a medication called midodrine can be very helpful.”
The three ‘Ds’ for fluid retention – diet, drinking, and daily weigh-ins
If you suffer from a lot of fluid retention, Dr Gibbs said it was important to cut out all the salt from your diet.
“Speak to your doctor about whether you should be on fluid restriction and how best to manage your diuretic medication (water tablets).
“Weighing yourself first thing every morning, after emptying your bladder, can be a helpful way to monitor your fluid retention,” he said.
“But as with everything, it’s always good to discuss these things with your doctor.”
Knowing your light chain levels is important
Dr Gibbs suggests patients with AL should be aware of their light chain levels at diagnosis and monitor these levels during each month of treatment. This involves a simple blood test.
“Patients with AL amyloidosis should ask their haematologist, ‘what was my light chain level at the start of treatment?’ Then ask what their light chains are every month during chemotherapy. Then you’ve got an idea of how you are responding to treatment.
“What we need is for that light chain level to decrease by at least 50-75%, absolute minimum, and optimally you’d want to decrease it by 90-100%.
“The main thing to bear in mind is that it often takes a few months to get a really good response,” said Dr Gibbs.
A simple blood test for earlier diagnosis
When Dr Gibbs was asked if he had a ‘holy grail’ of amyloidosis diagnosis and treatment, he said, “I would love a simple blood test that you could very easily do that would tell you, ‘yes, this patient is making amyloid’, or not, to help speed up diagnosis.
“The second holy grail would be a safe and effective medication that would remove the amyloid from the body quickly, easily, and without side-effects.
“Another unmet need is a more coordinated approach to patient care from various specialists,” he said.
“The AAN is collecting a series of amyloidosis experts in each organ speciality in each state to whom we can direct our patients when they say, ‘look, I’m struggling to find a good neurologist who knows about amyloidosis’ or ‘I’m struggling to find a good cardiologist’.
“One of the things the AAN is trying to do is to have a shared care model whereby we’re all working together; you have a haematologist who’s giving the day-to-day chemo, but you also have an amyloid specialist who’s supporting that local haematologist, to ensure you’re receiving the best supportive care as well as the best chemotherapy or TTR medication, and have access to clinical trials that may be beneficial.
“I do that a lot with patients in rural Victoria and Tasmania,” said Dr Gibbs.
What Dr Gibbs would do if he was diagnosed with AL amyloidosis
“I’d want to go and see a haematologist who was very experienced in amyloidosis, someone who is part of or closely connected to the AAN, or who attends amyloidosis conferences.
“If I received my treatment locally with an experienced haematologist, I’d still consider being seen at an AAN centre, at least initially, to ensure we had the correct type of amyloid diagnosed and the best supportive care mechanisms in place, and to see if there were any clinical trials that were applicable to me.
“I would want a specialist who sees me regularly for the organs that are involved with the amyloidosis. So, if I had, say, kidney, heart, and nerve involvement, I would want an experienced cardiologist, nephrologist, and neurologist looking after my heart, kidneys, and nerves respectively, who communicates regularly with my haematologist/amyloidosis specialist. However, I acknowledge that this can sometimes be a challenge.
“Being informed can be helpful. The Leukaemia Foundation produces a wonderful booklet (Amyloidosis – A guide for patients and families). So, I would read that booklet, and I’d review the AAN website, which is www.amyloidosis.net.au. There is the Leukaemia Foundation Facebook page for amyloidosis patients for support and it can be helpful for all sorts of different reasons. The ‘Amyloid Awareness’ cartoon on YouTube is also very interesting.
I would definitely attend any educational workshops about amyloidosis, to help understand the disease, and to keep up-to-date with the latest information and ideas about supportive care about this complex disease.”
The AAN is running Patient and Carer Educational Workshops on Saturday, May 8 in each capital city. For those unable to attend a face-to-face conference, the meeting in Melbourne will be live streamed and recorded online for later viewing. Check the AAN website www.amyloidosis.net.au regularly for details.
Amyloidosis in the future
Dr Gibbs is encouraged by the breakthroughs that have occurred over the last 10 years in the field of amyloidosis.
“Collaboration within Australia has made a huge difference in increasing awareness of the disease, leading to earlier diagnosis and treatment, and improved outcomes for patients,” he said.
“And international collaboration with research and clinical trials has accelerated novel drug discovery and accessibility within Australia.”
By working together and communicating regularly, Dr Gibbs believes everyone benefits.
If you are interested in making a donation in support of amyloidosis research, please contact the Leukaemia Foundation on 1800 620 420 or [email protected]
At 28-years-old, Jen was trialing all of the wellness crazes and ‘anti-cancer fads’ out there. Then she received a blood cancer diagnosis.
This story is a first-person account, contributed by a member of our wonderful community – all words are their own.
Jen will never forget the phone call that changed her life forever.
When I was 28 years old, I joined the club no one wants to be a part of. The Cancer Club. It was obviously a shock. I was the sort of girl that was trialing all the wellness crazes and “anti-cancer’’ fads. Alkaline foods, celery juice, going sugar-free, high-dose Vitamin C, essential oils, detox saunas – I’ve tried it all. At the end of the day, nothing could stop what was around the corner.
I’ll never forget the phone call that would change my life forever.
After a routine blood test, I was called just hours later by my GP with the shocking news. My white blood cell count was extremely high – worst case scenario it could be a form of blood cancer. It’s the sort of phone call that drains all the blood from your head and sucks the breath right out of you. What started as a perfectly normal day ended in a hospital bed, with my future uncertain.
Waiting for a diagnosis was by far the worst part of the experience. The only thing that stops the fear of the unknown is answers. Once we had an answer – acute lymphoblastic leukaemia (Philadelphia negative) – and a plan in place, the fear slowly dissolved. My old life ceased and my new full-time job as ‘Cancer Fighter’ began. Gone were the days of waking up, going to work and spending time with friends, family and my fiancé. Suddenly my life was all about pills, blood draws, chemo, radiation, neutrophils, chemo, x-rays, a stem cell transplant and of course, more chemo.
My treatment plan was long and comprehensive, but it was achievable. It was all about ticking off each step in the treatment plan. The physical side of treatment was half the battle, the rest was mental.
Being stuck in a hospital for six weeks straight, eating hospital food three times a day, getting prodded and poked, losing your hair and feeling like your body isn’t your own would get anyone down. The antidote for me, was talking to the cancer community.
When I first got that unforgettable phone call, my first contact was to one of my best friends who went through her own blood cancer treatment a year before. I was also put in contact with someone who did a similar trial to the one I was considering for my treatment. It was then that I decided to put my story on social media, in the hopes of connecting with more people going through something similar. I was so surprised with how many people I connected with. People all over the world who were going through the same diagnosis as me, or who had been through it and were on the other side. Beautiful people who reached out just to offer words of encouragement or advice for no other reason, but that they had been exactly where I was and knew the feeling.
What joining the ‘Cancer Club’ gave me
Throughout the challenges she’s faced, Jen’s grateful for the cancer community she calls friends.
The cancer community is full of empathetic, like-minded individuals and carers who know the struggle and who can offer their own experience or an ear to listen. It’s full of people who have been given another chance to enjoy the beauty this world has to offer, and they appreciate every moment of it. They’re willing to drop their plans to be with you if you need advice or someone to just sit with you. I know some people want to hide away from the world when diagnosed, but please know you’re not alone in your fight – we are all behind you and we’re here to talk when you’re ready. The Cancer club is the club no one wants to be in, but you’ll find it’s full of some of the greatest people you’ll ever have the pleasure of meeting.
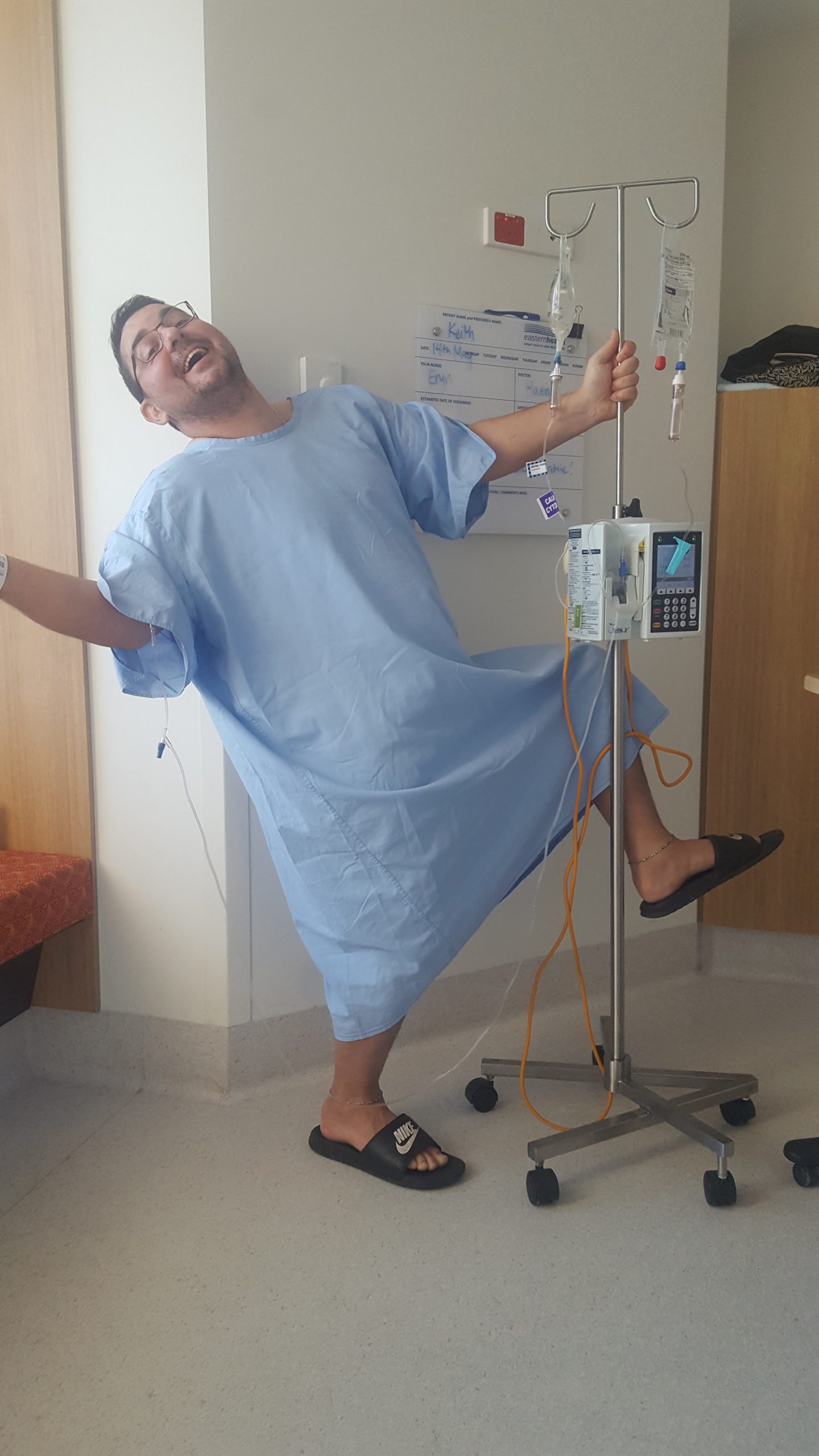
Diagnosed finally at 33 – Keith’s now in remission
Before going to Melbourne for his stem cell transplant, Keith Moore proposed to Felicity
Keith Moore was “ridiculously young” to be told he had AL amyloidosis and it took nine months before he finally got that diagnosis.
He’d just turned 33 and had begun an adult apprenticeship as a mechanical engineer after 14 years as a butcher.
“It was a totally different field; in something I love. It was fantastic,” said Keith, a professional speedway racing car driver.
But six weeks into that new job, Keith started getting ill. At first, he couldn’t get on top of a series of colds and flus. Then he got a painful burning sensation on the skin of his feet and chest, which felt on fire.
It took Keith Moore nine months to get a diagnosis: “I wish I’d had an earlier diagnosis.”
“I wasn’t sure what was going on, so we went to the doctor,” said Keith, now 36, of Murray Bridge, an hour north of Adelaide. His partner at the time, Felicity Czech who is now his fiancé, went along with him.
“My resting heart rate was abnormally high, about 90-95 beats per minute. That was a bit strange.
The doctor put Keith on an ECG machine and when this showed an abnormality, he was referred to a cardiologist.
Over the next nine months, Keith saw the cardiologist every four to six weeks and he also “bounced around between GPs, because they all seemed to think there was nothing wrong with me”.
“We were beyond frustrated as no one seemed to want to listen,” said Keith.
“It felt like I was being treated like a drug addict wanting drugs.”
“My heart rate was quite fast, my blood pressure was low, but the rhythm was fantastic, and I didn’t have a temperature.”
Keith developed other symptoms too, excruciatingly painful feet which meant, after working all week, he’d spend all weekend on the couch. He had a tingling feeling in his feet too and had trouble “going to the toilet”.
“I was sweating at night profusely,” said Keith and this meant getting up to change the sheets and his clothes during the night.
“I couldn’t sleep, then during the day I would sit down and fall asleep instantly for a bit, and numerous times I almost fell asleep driving the car home.”
He often got bloodshot eyes, vomited every day and night, was short of breath, and had shooting pains through his body, “like somebody just put a knife into me”. And he couldn’t drink alcohol.
But the biggest concern for Keith, an extremely fit, well-built guy who described himself as “80kg of pure muscle”, was the weight he lost over three months–20kg.
“I was nothing but skin and bone.
“I knew in the back of my mind there was something seriously wrong with me, but I’ve always been the sort of person that never got sick. I suppose I was in a lot of denial.
“I was sure they’d find out what was wrong, and I’d be right in a couple of weeks.
“It was a very strange and scary time.”
More than a decade earlier, when Keith was 21, he’d been diagnosed and treated for Wolff-Parkinson-White* syndrome.
“It’s quite a common thing. I’d had a heart ablation for that and was fine. Still to this day, I don’t have a problem with it at all.”
His cardiologist couldn’t work out what was wrong with Keith, so he referred him to another cardiologist. Keith had another “heap of tests” including an MRI scan of his heart. This picked up “some sort of infection” in his heart and he had a heart biopsy.
“We went in for the results six weeks later, and he said, ‘oh, you’ve got amyloidosis’.
“I had a brand new car and I took it for its first race, and won!” said Keith, a professional speedway racing car driver
“We had no idea what that was, and he didn’t explain it to us very well. He pretty much just said, ‘we can fix it with chemo, but I’m going on holiday, so I’ll see you in six weeks’.
“In my current state, I would not have lasted another six weeks,” said Keith.
He and Felicity said they “did a bit of Dr Google reading, which you really shouldn’t do”.
“What we read at that time was, ‘you have about 12 months to live’.
“That shot us down a fair bit, so we quickly made a GP appointment to double check our findings and we could tell by his face that this was not good, and he said we should talk to our families and make a plan.
“I had to tell my young children that Dad was going to go in about a year. It was horrible,” explained Keith who has three children, now aged 17, 13, and 11.
Scrambling, and feeling like everyone had already given up on him, Keith jumped on Facebook seeking help. He found a support group and immediately posted, “this is what I’ve got. I’m quite scared. I don’t know what to do”.
“A lady rang me. She gave me a bit of a back story on amyloidosis. Her husband had passed away 10 years prior.
“She said, ‘get on to this guy, Simon Gibbs, he’s Australia’s leading expert’.
“I emailed Simon the next day and said, ‘please help. I’m 33. This is what I’ve got’. It was a Thursday. He rang me straight away and said, ‘can you come to Melbourne on Monday?’.
“I said, ‘yep, we’ll be there’, so my father and my partner booked tickets and we were off on the plane to Melbourne on Sunday, and on Monday, Simon took one look at me and stuck me in hospital to start chemo the next day.”
“We thought this would be a 24 hour round trip!”
At the time, Keith didn’t realise how close he was to dying.
“I didn’t want to have chemo and when we asked about other treatments available, Simon’s reply was, ‘I don’t think anyone wants to have chemo’.
Keith, dressed for the speedway, with Felicity and his two youngest children
“He pretty much said, ‘if I don’t give you chemo, you won’t last two weeks’; that’s how frail I was.”
“Simon told me amyloidosis had come a long way in 10 years. It had been a death sentence, but today more people are surviving with new treatments, and CAR T-cell therapy is coming which is meant to be the new greatest thing.
“Amyloidosis is quite rare. Most people who have it are 60, 70, 80 years old, so to find it in somebody 33 years old is just ridiculous.”
“We have a very good relationship, Simon and me. I said to him, ‘look, no bulls..t. Don’t sugar coat it, tell me how it is. I need to know what’s happening. If you do that, I’ll guarantee I’ll get through it’,” said Keith.
In Melbourne, Keith started chemotherapy and arrangements were made for him to see a local haematologist back in South Australia, and a week later, he flew home and continued the medication Dr Gibbs had given him.
“I finally got to see the haematologist [in Adelaide] two weeks later and she did the same thing, she took one look at me and stuck me in a hospital. She didn’t think I had a chance of surviving, but Simon’s response to her was – ‘he’s young, he can do this’.”
Keith continued the treatment for six months, then went back to Melbourne to see Dr Gibbs.
“He said, ‘right, another three months-worth of treatment’.
The issues Keith had during his 12 rounds of VCD**, began in the first month when he was rushed to hospital in a lot of pain, having not had a bowel movement for five days, then during the last cycle of chemo, he got a urinary tract infection and a cold.
Keith loves a bit of salt but people with amyloidosis need to limit their salt intake so he paid the price with a lot of fluid retention in his legs and in his chest, which affected his breathing.
“Other than that, I didn’t get sick and stayed completely healthy,” said Keith.
“We got the light chains down as far as we could until the treatment stopped working, then he [Dr Gibbs] said, ‘right, you’re fit, you’re healthy, you’ve put on weight’–I was back to 80kg–‘let’s do it’.
Keith went back to Melbourne to have his stem cells collected in the leadup to having an autologous stem cell transplant.
“While I was in hospital, Simon had a patient who had just had a stem cell transplant come and talk to me. He was fit and healthy and back to work. “He was actually a mechanical engineer like I am, which was fantastic.
“We related to each other a fair bit. He was an older guy, in his 50s, which is still quite young for amyloidosis. We had a great chat and he told me what he went through.
“That was really helpful… to see it can be done. That was a big turning point.
“He was pretty confident I’d be fine, which was the hope we needed.
Keith larking about during treatment
Three weeks later, at the end of May 2019, Keith had his transplant at Box Hill Hospital and a week later Felicity had to return home to the children and work.
During his five weeks in hospital, Keith almost died from an infection that ended up being associated with his PICC line, which was removed.
Then, after spending another week in Melbourne, at a hotel***, “to double check nothing was going to go wrong”, Keith and his dad took two days to drive home.
“I spent another seven weeks recovering at home before I went back to work full-time.”
“My family, the doctors, everyone, all told me off and said I went back to work too early,” said Keith, but he was determined.
“My work was fantastic. When I explained what was happening to me [after Keith’s diagnosis] and that treatment would take 12 months, they organised for my apprenticeship to continue and gave me 12 months off work. My job would be waiting for me when I returned.
“I had almost 12 months off to the day.”
Since returning to work in August 2019, Keith has gone from strength to strength.
During his transplant, Keith had problems keeping his food down, and for nine months he continued to struggle with nausea. But the peripheral neuropathy in his feet, caused by the amyloidosis, has “pretty much gone”, along with his former fatigue, and now he’s “the perfect weight.
“I eat fine. I drink fine. I go to the toilet fine. My heart rate has come down.
“I’m probably 95% back to where I was. I do have a little bit of nerve damage, but not too much. I still get a little bit tired every now and then. I have to make sure I drink water consistently and am constantly hydrated. And my vision was affected. Now I wear glasses all the time.”
Keith’s not on any maintenance medication and his bloods are in the normal range.
“It’s a bit of a miracle to be honest.”
During December 2019 and January 2020, Keith’s light chains started to rise again, and arrangements were put in place for him to have lenalidomide (Revlimid®). This took some time, and when Keith finally got access to the treatment, in April, COVID-19 had struck.
“When I called Simon, he said it was probably not a good idea to take lenalidomide as it would lower my immune system. He decided to do another test on my light chains. If they were high, I would have to take the lenalidomide, but if they were low, we’d hold off and keep an eye on them.”
It turned out, however, that Keith’s light chains had gone down on their own.
“They went back to normal and have been normal ever since,” he said.
“I am currently in remission. We try not to use that word, and Simon even dropped the word ‘cured’ once. He said, ‘we don’t like to say it, but we think you’re cured, but we won’t say that ever again’.”
Keith has a blood test every six to eight weeks “just to make sure”, and everything has been in the normal range for 12 months now, with no maintenance therapy whatsoever.
Keith Moore with Dr Simon Gibbs: “I was Simon’s youngest AL amyloidosis patient”
Reflecting on his experience with amyloidosis, Keith said, “it is hard to talk about. It’s quite scary what we went through and the fear of it coming back is still there, and that will never go away unfortunately”.
“We know it’s there. We know that one day we’ll probably have to deal with it [amyloidosis] again.
“We try to put it out of our minds and just keep on moving and trying to live a normal life,” said Keith, who still has more stem cells “on ice, waiting for me, if I ever need them again”.
“We’ve got a whole new perspective on life. We do things differently; we don’t take things for granted now. We take the time to appreciate everything.
“We don’t go out and party hard like we used to, we sit back and relax and look at the world now instead.
“We spend more time with the family, try not to work as many long hours, don’t worry about the small stuff.
“We’re getting married as well.”
“I proposed before I went over to Melbourne for the stem cell transplant,” said Keith, and the wedding is being planned for next year.
Keith, who had to give up racing for three years due to his illness, recently returned to the speed track. Some of his friends took his race car away to re-do it.
“Some of my friends took my car away to re-do it, so I didn’t have to worry about it.
“While I was going through treatment and without me knowing, they got together with my friends, family and sponsors, and built me a brand new top-of-the-class race car,” said Keith.
Unfortunately, it didn’t quite get finished–the business of the people who took on the project, took off and they ran out of time. So when Keith got back from Melbourne, he picked up where they left off.
“I had a brand new car and I took it for its first race, and won!”
He put a myeloma ribbon decal on the bonnet and under the words, “Amyloidosis awareness”, to promote the two diseases.
“I also have myeloma,” said Keith.
“When we went over to Melbourne for the transplant, the Leukaemia Foundation helped out, paying for a bit of accommodation in the hotel while I was there, which was fantastic.
“And we got a heap of information from them, which was really good. They posted that out to us,” said Keith, and in 2019 he took part in the Leukaemia Foundation’s Light the Night event.
When it comes to advice for others with amyloidosis, Keith is adamant: “Don’t read the internet. That’s number one”.
“Although we did Google ‘the best diet for someone with amyloidosis’. The Mediterranean diet was recommended, and we stuck to it – fish twice a week and lots of berries.
“For nine months, I ate a punnet of strawberries every single day.”
Keith wishes he’d had an earlier diagnosis.
“The hardest part was not knowing and not getting answers for such a long time,” he said about those nine months.
“We hope this story helps others by lifting the spirits of those suffering with amyloidosis and myeloma and those caring and supporting them,” said Keith.
“And hope lies with this story, which proves that its ass can be kicked.”
* Wolff-Parkinson-White (WPW) syndrome is a rare congenital heart disorder involving irregularities in the electrical system of the heart. Treatment may involve medication, a procedure known as ablation, or, in rare instances, an electric shock may be used to restore a normal rhythm.
** VCD is a combination of bortezomib (Velcade®), cyclophosphamide and dexamethasone.
*** The Leukaemia Foundation helped out by paying for this accommodation.
Approval for Australian CAR-T therapy manufacturing site to deliver faster access to innovative treatment for patients with aggressive blood cancers
Friday February 12, 2021
Breakthrough CAR-T therapy is now another step closer to being manufactured in Australia for the first time, allowing faster access to the innovative treatment for eligible Australians with life-threatening blood cancers.
The Therapeutic Goods Administration (TGA) has now granted approval for Cell Therapies Pty Ltd to commence commercial manufacturing of Kymriah® (tisagenlecleucel) in its manufacturing facility at Melbourne’s Peter MacCallum Cancer Centre.
The approval, announced today, means the site becomes the first and only approved commercial manufacturing hub for CAR-T therapy in the country for both clinical trial and non-clinical trial patients, securing supply of the state-of-the-art treatment onshore.
Primarily used to treat blood cancers, CAR-T therapy is a once-off, individualised treatment that uses the patient’s own reprogrammed T-cells to fight their cancer. The treatment involves extracting a patient’s own immune cells and genetically re-engineering them before infusing them back into the body.
For some patients CAR-T therapy will lead to a cure. Where other patients relapse, the therapy will be used as the mechanism to lead to remission to enable a stem cell transplant as the next line treatment option.
Today’s announcement comes after the TGA last year approved the use of CAR-T therapy in Australia for some Australians with aggressive lymphoma and leukaemia, and also follows an $80 million investment by the Federal Government to establish the Peter MacCallum Cancer Centre as a new base for CAR-T therapy and other immunotherapy clinical trials.
Leukaemia Foundation CEO Chris Tanti applauded the approval, saying the organisation recognised CAR-T therapy as a potential gamechanger in the treatment of patients with aggressive blood cancers and local manufacturing means improved security of supply and greater local capacity to deliver CAR-T therapy to these Australians.
“Until now, Australians with blood cancer receiving this therapy had to have their cells shipped overseas to the USA, so approval for this advanced manufacturing process to remain onshore means that from start to finish, this treatment can be produced and delivered here, and therefore more efficiently,” he said.
“For Australians diagnosed with aggressive blood cancers, every day counts in their treatment journey, and today’s announcement represents faster and easier access to a potential lifeline for some of these patients right here, right now, which is an incredible win.
“Targeted treatments, precision medicine and immunotherapies are changing the face of how we tackle blood cancer in this country. The Leukaemia Foundation is committed to driving development in this area and strongly supports any initiatives to fast-track and increase access to innovations like CAR-T therapy as we set our sights on the blood cancer community’s shared vision to achieve zero lives lost to blood cancer by 2035.”
Subsidised access to CAR-T therapy is available for eligible Australian patients with relapsed or refractory Diffuse Large B Cell Lymphoma (DLBCL), and eligible paediatric or young adult (up to 25 years) patients with relapsed or refractory Acute Lymphoblastic Leukaemia (ALL).
To date, more than 100 Australians have been treated with CAR-T therapy across clinical trials and commercially available therapy in seven treatment sites spanning Victoria, New South Wales, Queensland and Western Australia.
The Leukaemia Foundation has committed to supporting patients with free emotional and practical support including accommodation close to each treatment centre for the full duration of the treatment period while undergoing CAR-T therapy.
“Whether you live in a capital city or a rural township, access to new blood cancer therapies is a critical factor influencing survival and it’s our priority to ensure all Australians living with a blood cancer have equal access to the best treatment possible, no matter where they call home,” Mr Tanti said.
END
Subscribe to Leukaemia Foundation news and updates
Nearly 16 years since her diagnosis, Emma Ingster, now 30-years-old, reflects on being diagnosed as a teenager and the challenges of navigating a health system where she often felt invisible.
“I’d never been to hospital or had any illnesses besides a common cold before being diagnosed,” remembers Emma.“I was in year nine and spent most of my spare time outside of school writing books and poems.”
As soon as Emma was told what she had was very serious and she could die, she entered what she refers to as “the second half of my life”.
Emma on the last day of her treatment cycle before beginning maintenance.
“Nothing was ever the same after that,” she said.
“My mum was devastated, my much younger sister was too young to fully comprehend what was happening, and my dad kept focused on work to dealwith my diagnosis.”
With no proper treatment facilities or protocol available to treat her as an adolescent, Emma wasn’t able to have treatment in her hometown of Canberra.
“The doctors told me if I received an adults’ treatment protocol, my overall survival rate would drop significantly,” explains Emma. “My best chance was the Sydney Children’s Hospital in Randwick.”
Emma and her mother, Esther were flown by Care Flight to Sydneywhere her oncologist confirmed she had a rare form of T-Cell ALL, only documented in a handful of people worldwide.
“My oncologist consulted with her peers in America,” explains Emma.“They agreed to implement a nine-month intense chemotherapy protocol followed by and an 18-month maintenance treatment as opposed to six months of chemo and three months of radiation.
“My mum and I travelled back and forth to Sydney for nearly a year for my treatment.”
She nowlooks back on this with disappointment and hopes that things can be improved for teenagers facing a diagnosis.
“It upsets me a lot to this day that adolescents still fall into an invisible category.”
“Knowing my own hometown could not cater for my physical treatment needs due to my age was disappointing and I would have felt more supported had better systems for teens with cancer been in place.
“I was first admitted onto an adults’ oncology ward where I was surrounded by elderly people with no one to talk to.
“I was then transported to a children’s ward covered in posters of the Bananas in Pyjamas. I was too physically sick to attend any camps; my only contact with teens was with a few others who had been diagnosed around the same time as me
“Things have improved somewhat, but there is still the invisibility of being between child and adult that means adolescents are not always supported to their exact needs.”
Emma missed a year and a half of school due to her treatment and found it difficult to return to her pre-cancer life.
Emma and Joe celebrating Emma’s 15 years remission from leukaemia.
“Facing your own mortality much sooner than you expected challenges how you live your life from that point onwards,” said Emma.
“Many of the things I had once considered important to me weren’t anymore and the things that were now important to me were not necessarily on the minds of other people my age, so I often felt a lot older than my peers.
“Gradually, I learned to accept I would be different, but that I could still enjoy being young and having fun.
“I also struggled for many years to figure out my place in the world-the pressure of surviving made me want to live perfectly.”
On the 4th May 2020, Emma celebrated fifteen years remission from leukaemia with her family including husband, Joe who is one of her former classmates.
“Support from people I love, even support from strangers, has impacted my life for the better and it continues to help in my healing,” said Emma.
“I am very lucky that I have no physical side effects from my treatment to date.
“I was even told by my GP that my fertility is normal, which was a nice surprise. Unfortunately, leukaemia has meant I suffer with anxiety on a regular basis, but I am learning self-care to combat that.”
Emma offers this advice to others going through a diagnosis:
“You are in a new world so always ask questions about anything you don’t understand and don’t be afraid to tell the doctors/social workers/counsellors what you need or what you would like to see improved for yourself and others,” she said.
Emma plans to finish her Masters of Teaching later this year.
“Some family members and friends may greatly disappoint you; it will hurt, but you are better for knowing who those people are and investing your love and energy into the others who are there for you.
“Think about what you want to do in the future, but not so much that you forget to exist in the present.”
“As you heal, never forget to take pleasure in the small things like a good cup of coffee, beautiful nature, a hug from someone who loves you.
“The bigger things will come later. Don’t expect perfection but look forward to good things happening again because they will.”
Emma is a keen writer in her spare time andis about to finish her Master of Teaching.
“My aspiration is to continue to write and publish my novels and my memoir someday,” said Emma.
“As for the rest, I want to continue working in education and also fundraise and volunteer when I can. If I become a mum someday, I’ll aspire to teach my children that they are stronger than they could ever possibly know.
“Oh, and I hope to live to be a sassy one-hundred-year-old woman!”