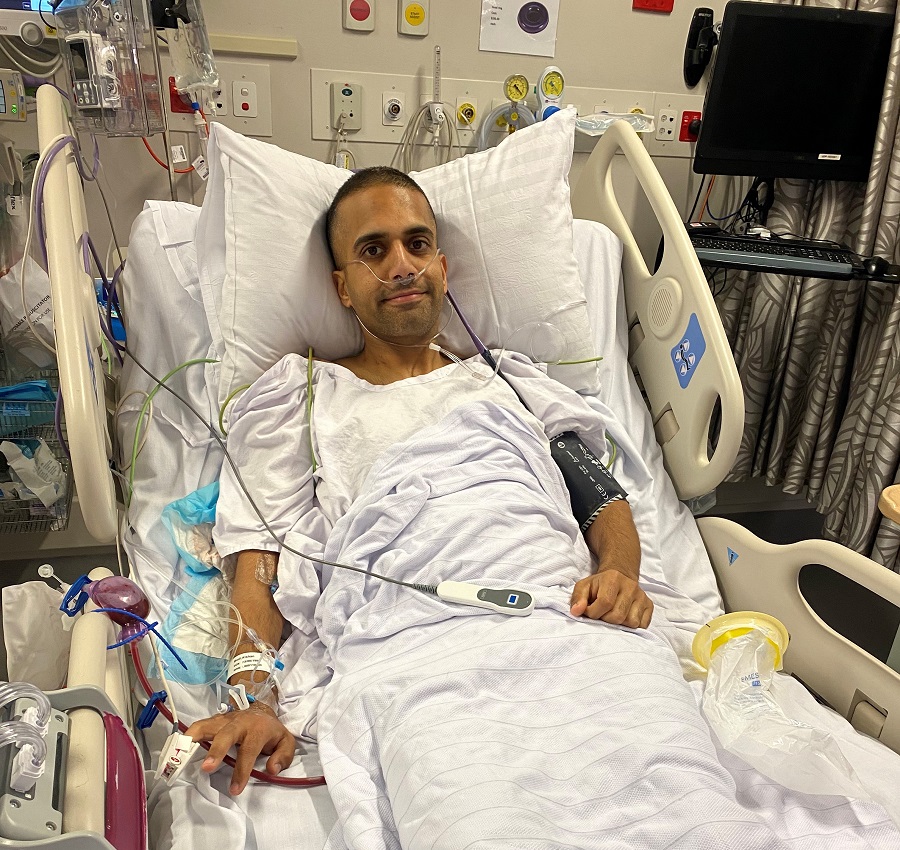
“Live life to the fullest”: Josh Gourlay opens up about returning to work and new beginnings after blood cancer
While a shock blood cancer diagnosis put 34-year-old Josh Gourlay’s life temporarily on hold, it also presented him with a new mindset, and new opportunities.
- Acute lymphoblastic leukaemia
- Treatments and side-effects