Light the Night in your own special way this year
Learn how this beautiful blood cancer community event has been re-imagined in 2020.
- Our supporters

Learn how this beautiful blood cancer community event has been re-imagined in 2020.
Deb Henderson shares her story to help us mark Blood Cancer Awareness Month. Deb has blood cancer and is also a member of the Blood Cancer Taskforce.

Blood cancer survivor Sharyn Polce has experienced first-hand the ‘postcode lottery’ of treatment

This Blood Cancer Awareness Month, find out how you can help people with blood cancer get the treatments they need.

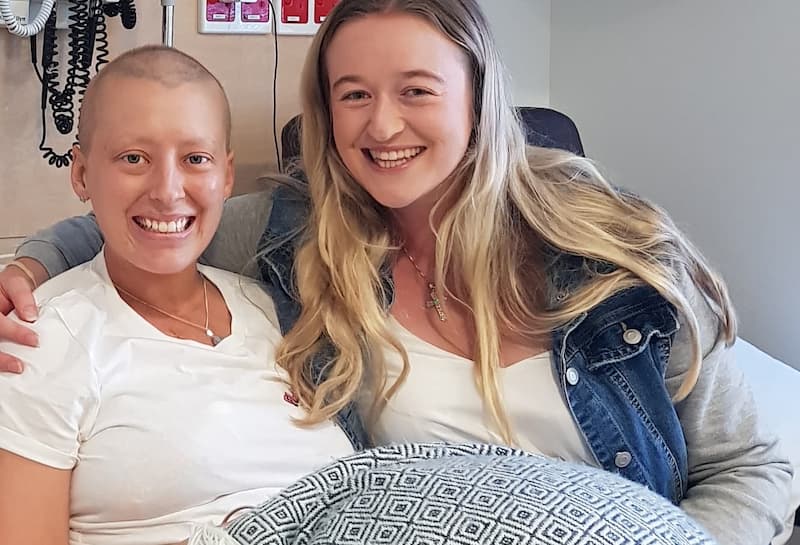
Courtney and Kate's participation in Dry July helped fund our nation-wide network of Blood Cancer Support Coordinators.
We're working closely with the Fini Group to support the outcomes of a new health and knowledge precinct.

Jennie and Russell Wigginton were planning for baby number two, when Jennie was diagnosed "out of the blue".

Professor David Ross discusses the "massive explosion" of recent clinical trial activity in MPN.

Dr Liesl Butler is investigating the gene mutations and biological pathways that lead to the development of MPN.
Through the help of our supporters, we've been able to create an e-learning course for carers.
